gmx_Hbond
本模块依赖GROMACS进行氢键的计算,包括氢键的数量、时间占有率、氢键形成的平均距离和角度等。
使用本模块前请注意前置处理已经完成!
Input YAML
- gmx_Hbond:
group1: Protein
group2: ZIN1 #Ligands # connot start with a number
top2show: 6
only_calc_number: no
gmx_parm:
b: 50000
e: 90000
group1和group2是需要计算氢键的两个原子组,如果需要计算同一组原子内的氢键的话,两个组可以写成一样的。需要注意的是,组名不能是数字开头的,例如1ZIN这样的组名,会导致GROMACS识别到错误的组。
top2show是指展示前面多少个占有率最高的氢键数量,默认是6,可以根据需要调整。
如果只需要计算氢键数量,或者当预估的氢键数量特别大时(例如计算蛋白质和水的氢键),可以将only_calc_number设置为yes,也即只计算氢键数量,而不计算其他参数。
gmx_parm参数下面可以写一些gmx的参数;由于此模块涉及到gmx hbond、gmx distance和gmx angle命令,s所以这里只允许6个参数:a、r、da这三个参数只会被连接到gmx hbond命令中,而b、e、dt参数会被连接到全部三个命令中;其余的参数都将被忽略。
Output
首先DIP会可视化gmx hbond输出的氢键数量图和氢键占有率图:
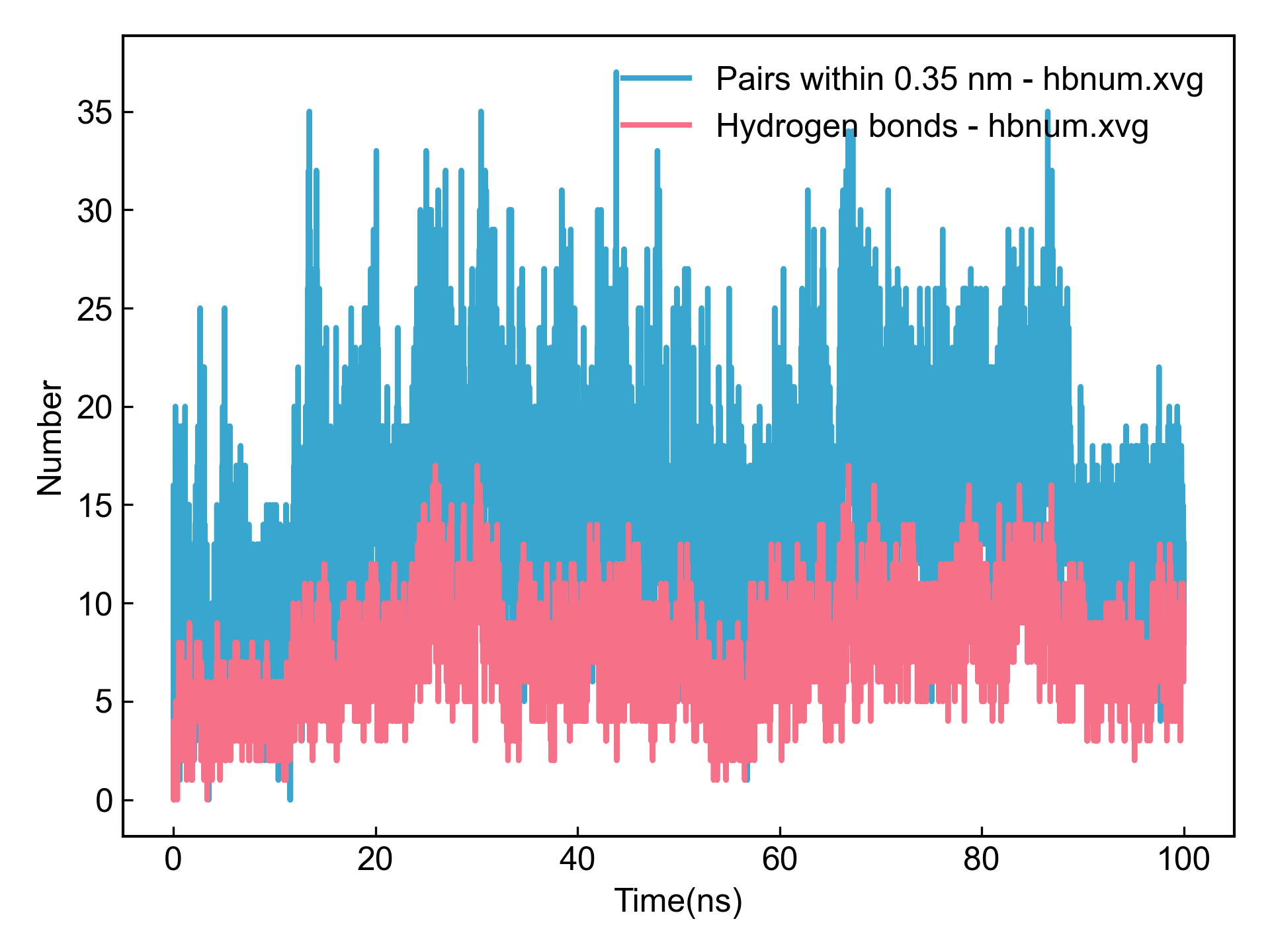
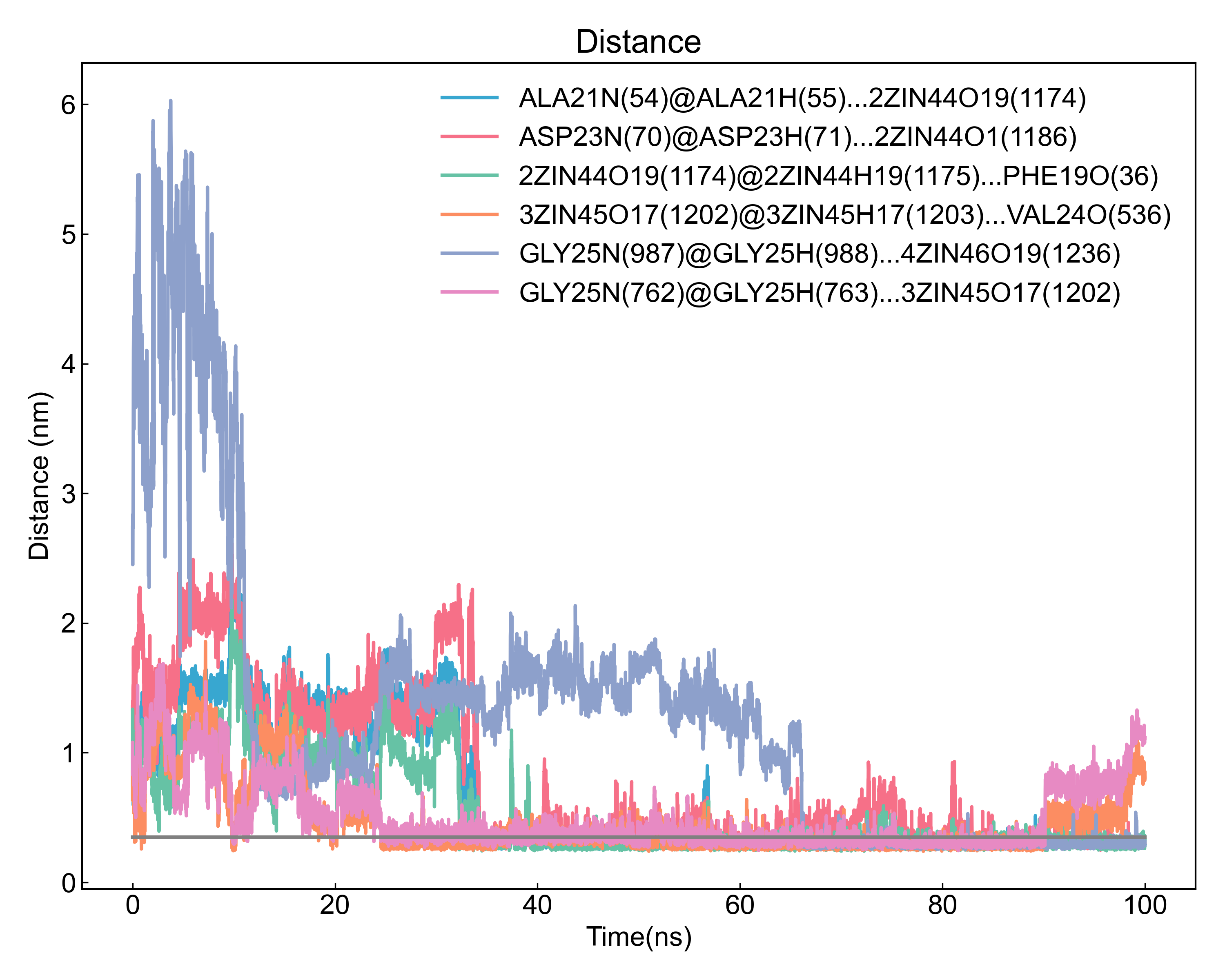
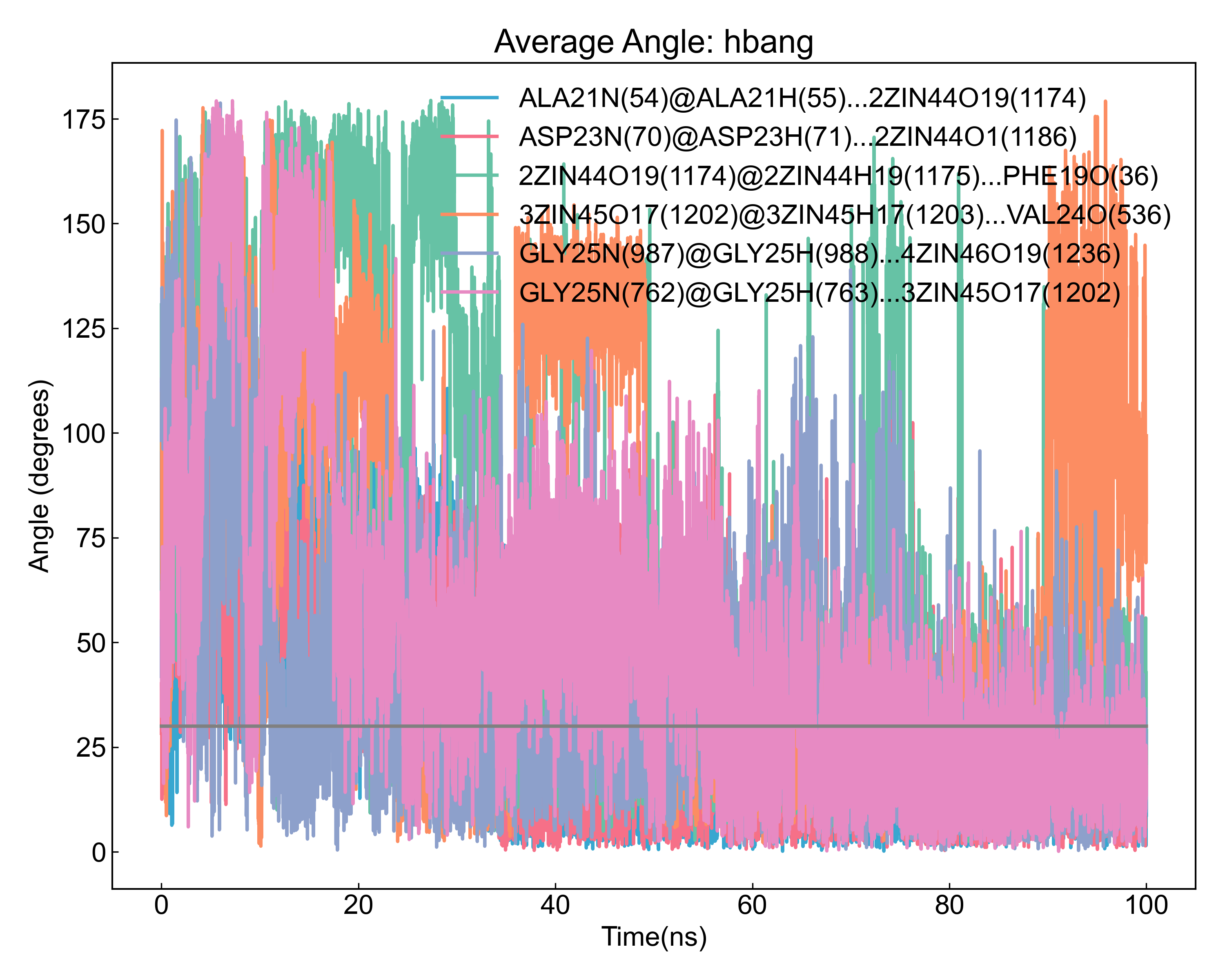
之后DIP会对检测到的占有率最高的几个氢键,计算其距离和角度随时间的变化,如图:
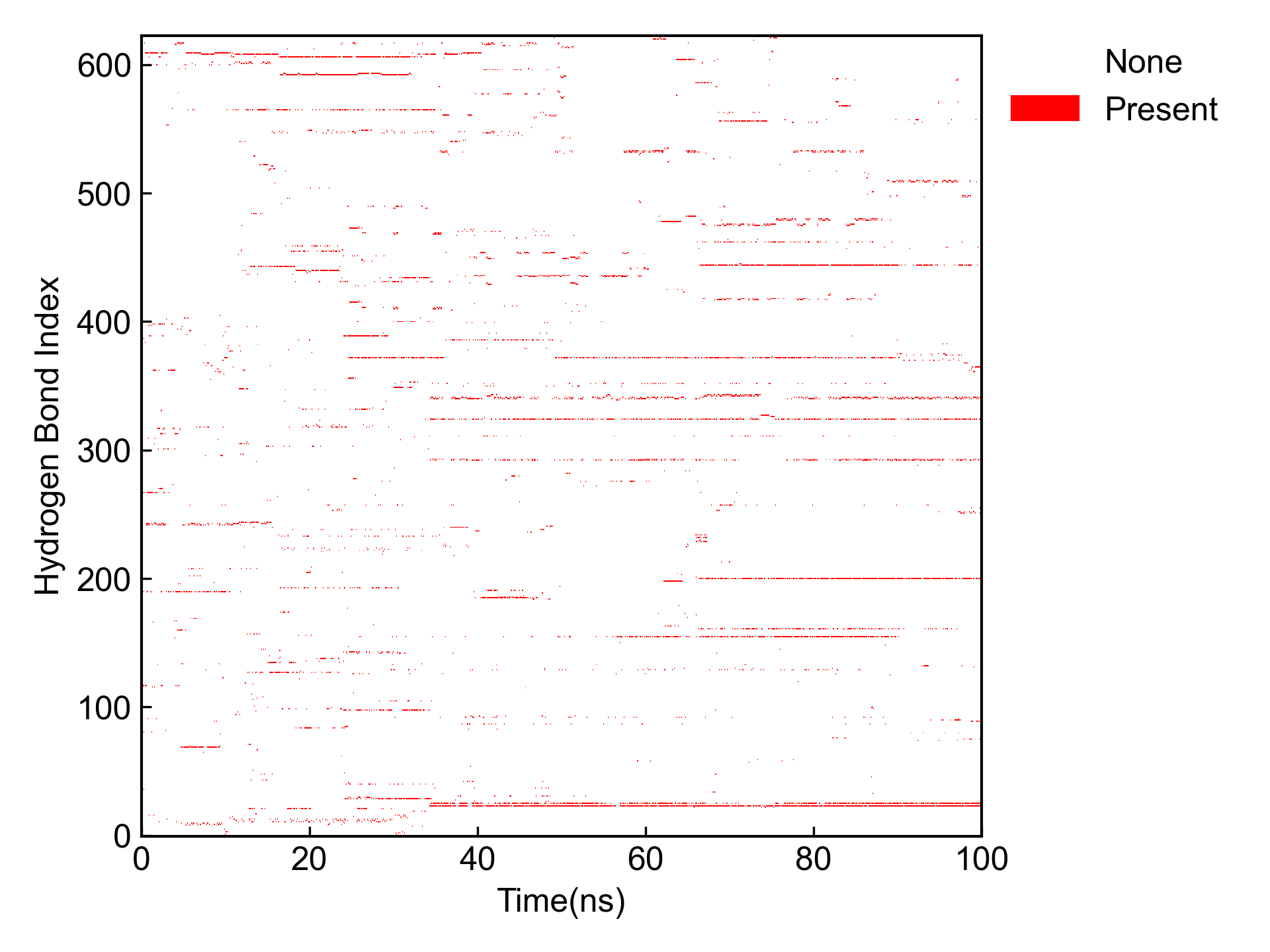
同时还会对占有率最高的几个氢键进行占有率的可视化:
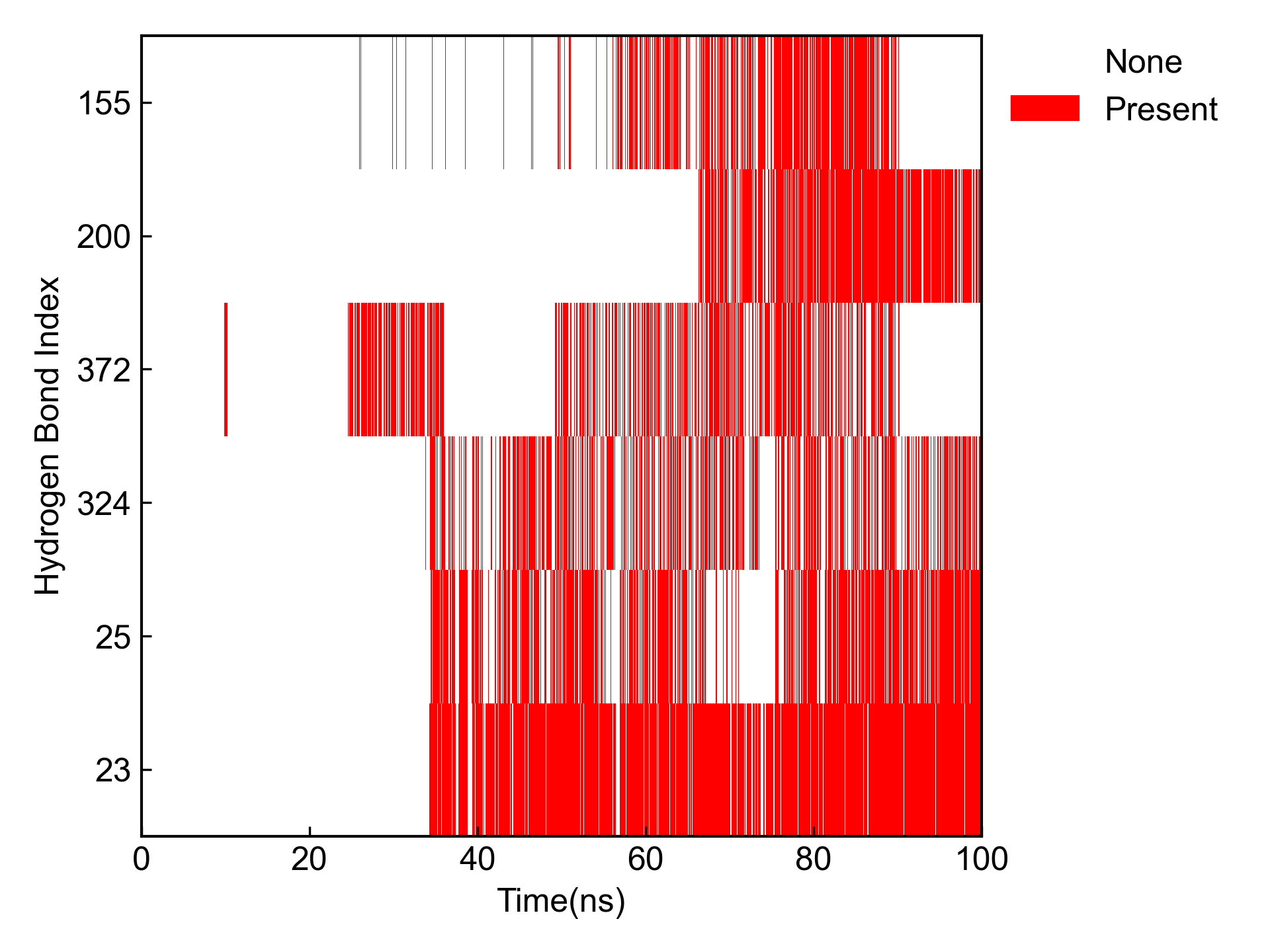
DIP会统计所有氢键的时间占有率、形成了氢键的平均距离和平均角度,并输出到csv文件中:
id,donor@hydrogen...acceptor,occupancy(%),Present/Frames,Distance Ave(nm),Distance Std.err(nm),Angle Ave(deg),Angle Std.err(deg)
0,LEU34N(383)@LEU34H(384)...1ZIN43O17(1140),0.02,1/4001,0.3030,0.0000, 9.38,0.00
1,MET35N(392)@MET35H(393)...1ZIN43O12(1133),0.02,1/4001,0.3280,0.0000, 17.28,0.00
2,MET35N(392)@MET35H(393)...1ZIN43O19(1143),0.05,2/4001,0.3050,0.0283, 23.95,3.43
3,LEU34N(608)@LEU34H(609)...1ZIN43O12(1133),0.10,4/4001,0.3300,0.0097, 20.82,7.01
4,LEU34N(608)@LEU34H(609)...1ZIN43O19(1143),0.07,3/4001,0.3103,0.0295, 22.75,2.69
5,MET35N(617)@MET35H(618)...1ZIN43O19(1143),0.07,3/4001,0.3227,0.0225, 18.74,9.73
6,LEU34N(833)@LEU34H(834)...1ZIN43O17(1140),0.02,1/4001,0.3160,0.0000, 29.34,0.00
7,1ZIN43O12(1133)@1ZIN43H12(1134)...GLY33O(157),0.87,35/4001,0.2846,0.0206, 15.18,7.49
8,1ZIN43O12(1133)@1ZIN43H12(1134)...ILE31O(368),0.10,4/4001,0.3058,0.0311, 23.62,6.99
9,1ZIN43O12(1133)@1ZIN43H12(1134)...GLY33O(382),3.77,151/4001,0.2892,0.0214, 15.45,6.85
氢键名称由[供体@氢…受体]组成, 每部分的命名含义如下:残基名字、残基编号、原子、括号里面是原子编号。
References
如果您使用了DIP的本分析模块,请一定引用GROMACS模拟引擎、DuIvyTools(https://zenodo.org/doi/10.5281/zenodo.6339993),以及合理引用本文档(https://zenodo.org/doi/10.5281/zenodo.10646113)。