gmx_Mdmat
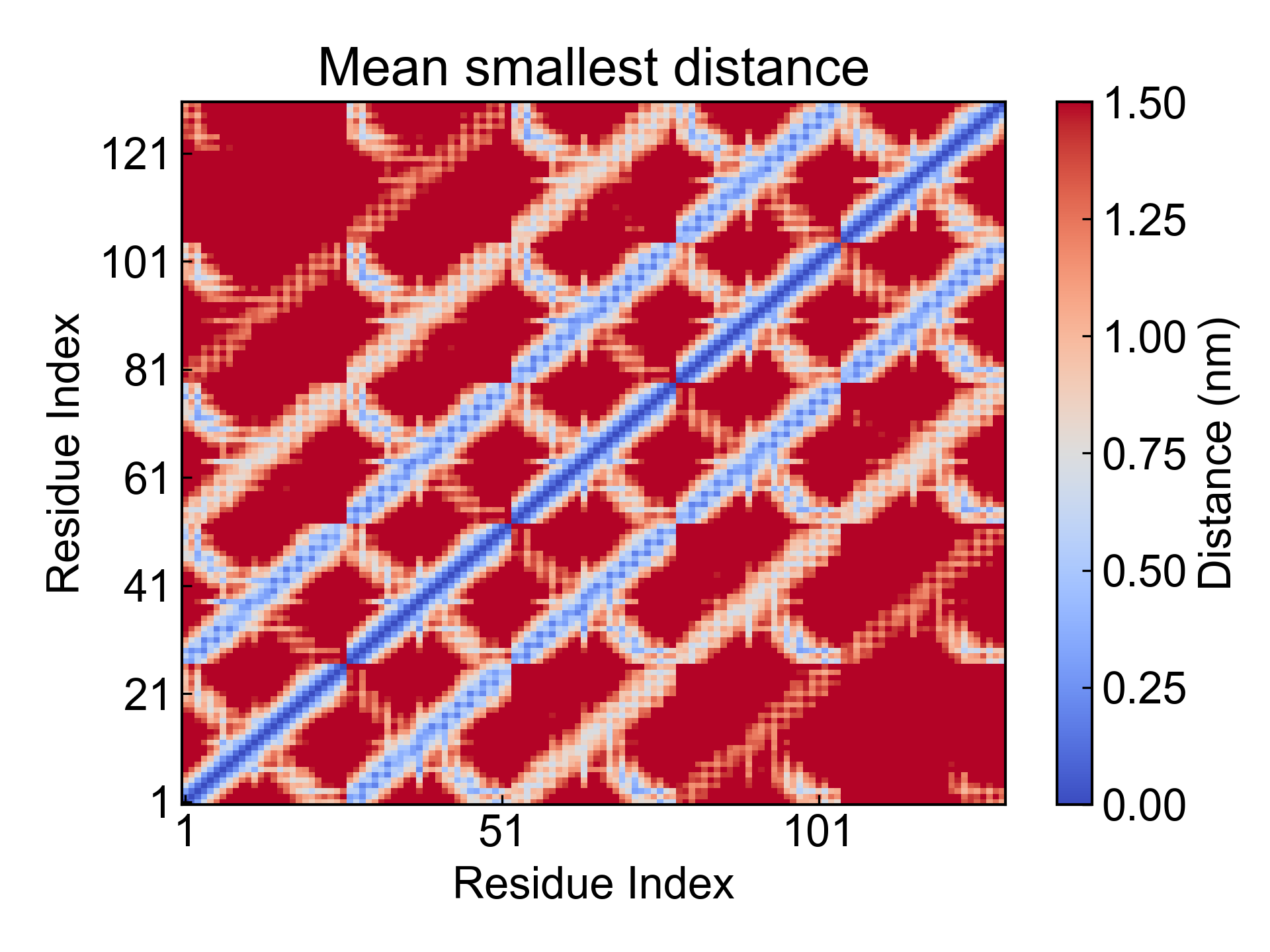
此模块利用GROMACS计算残基间的最短距离矩阵,也可以称之为残基接触矩阵。
使用本模块前请注意前置处理已经完成!
Input YAML
- gmx_Mdmat:
group: Protein
gmx_parm:
t: 1.5
只需要确定需要计算的组就可以了。也可以通过gmx_parm参数来设置一些额外的参数,如这里设置了距离截断-t 1.5。DIP默认会调用-mean -no两个输出参数,因而这俩个不需要在这里添加。
Output
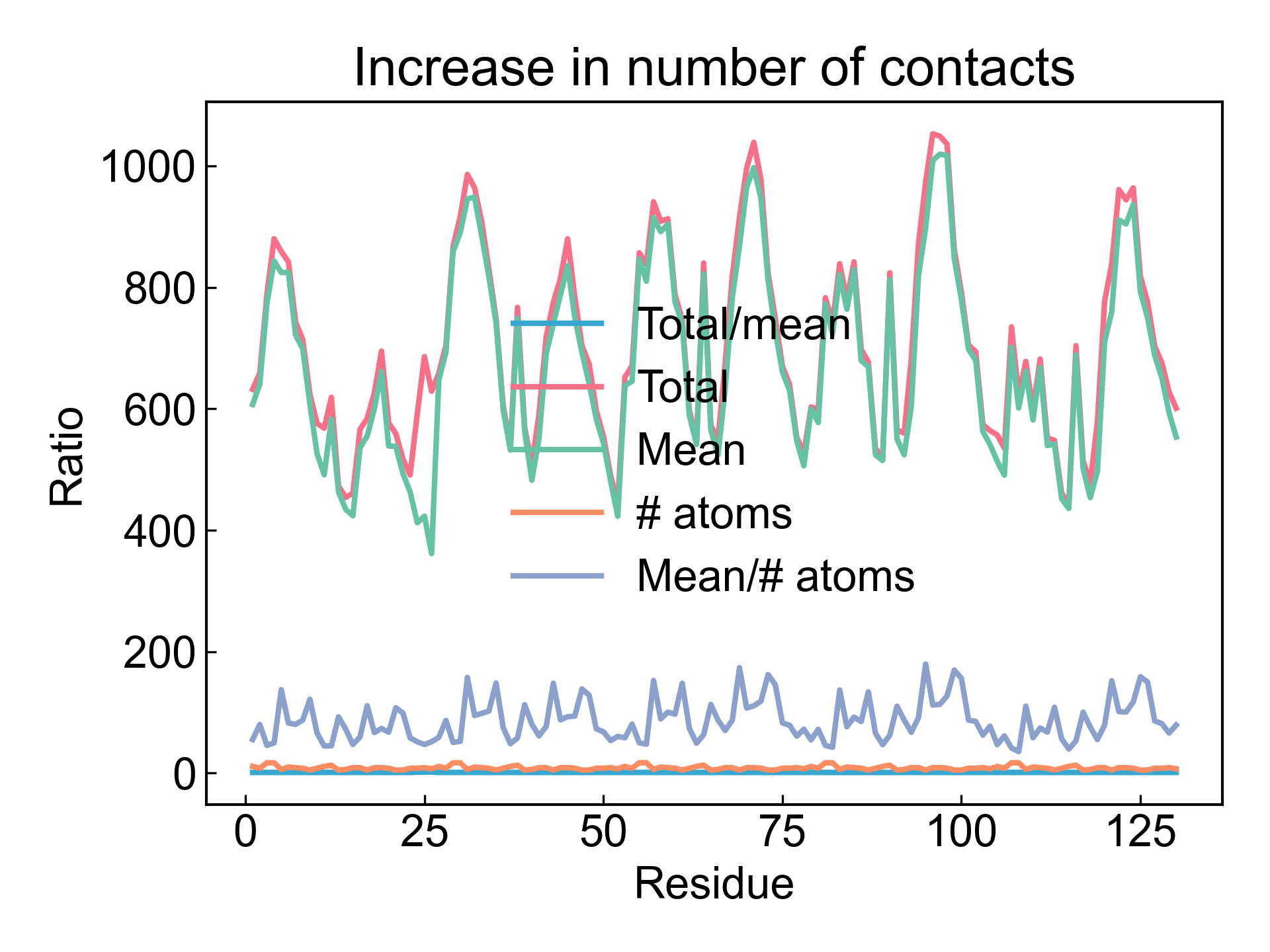
DIP会将产生的平均的残基接触矩阵可视化,以及绘制接触数量随时间变化的折线图:
References
如果您使用了DIP的本分析模块,请一定引用GROMACS模拟引擎、DuIvyTools(https://zenodo.org/doi/10.5281/zenodo.6339993),以及合理引用本文档(https://zenodo.org/doi/10.5281/zenodo.10646113)。